Still a good boy nearly 100 years after historic sled run, Balto has now helped scientists explore the genetics of working dogs and demonstrate the power of comparative genomics


The sled dog Balto has been celebrated in books and movies for his role in delivering desperately needed diphtheria antitoxin to Nome, Alaska, in 1925. Now, his DNA has enabled scientists to explore the genetics of 1920s sled dogs in Alaska and understand how they compare to modern dogs.
Scientists at UC Santa Cruz sequenced Balto’s genome as part of a large collaborative effort in comparative genomics leading to several papers published in the April 28 issue of Science. For the Balto study, the UCSC team extracted DNA from tissue samples of Balto’s taxidermied remains, provided by the Cleveland Museum of Natural History, and worked with colleagues at Cornell University and other institutions to investigate his ancestry and genetic traits.
“Balto’s fame and the fact that he was taxidermied gave us this cool opportunity 100 years later to see what that population of sled dogs would have looked like genetically and to compare him to modern dogs,” said Katherine Moon, a postdoctoral researcher at UC Santa Cruz and first author of the paper on the team’s findings published in Science.
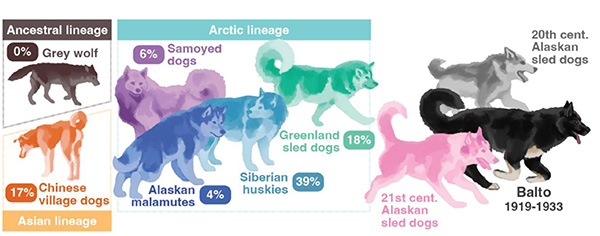
The researchers found that Balto shared just part of his diverse ancestry with the Siberian husky breed. He belonged to a population of working sled dogs that were more genetically diverse than modern breeds and differed not only from today’s Siberian huskies but also from modern Alaskan sled dogs. The study found evidence that his population was genetically healthier than modern breeds and carried gene variants that may have helped the dogs survive their extreme environment.
“Balto came from a population of working dogs that were different from modern breeds and were adapted to harsh conditions,” said coauthor Beth Shapiro, professor of ecology and evolutionary biology at UC Santa Cruz and a Howard Hughes Medical Institute investigator.
The analysis of Balto’s genome involved comparing it to a dataset of 682 genomes from modern dogs and wolves, as well as an alignment of 240 mammalian genomes developed by the Zoonomia Consortium, which was the foundation for most of the new comparative genomics studies published in the special issue of Science.
“We were able to take advantage of both the Zoonomia alignment and the huge amount of work that has gone into collecting the genomes of dogs,” said Shapiro, who is a member of the Zoonomia Consortium.
She explained that a key innovation behind these new studies is the ability to align the genomes of hundreds of species so that corresponding positions in different genomes can be compared. Comparative genomics can then reveal DNA sequences that are the same across species, having remained unchanged over millions of years of evolution—an indication that these are important parts of the genome where mutations could be harmful.
“A gene that’s on one chromosome in us is on a completely different chromosome in another species,” Shapiro said. “You need a tool that can line them up so you can see which parts of these genomes are the same and which are different. Without that it’s just a bunch of genomes of species that are very divergent.”
The genome alignment tool that made this possible was developed by researchers at the UC Santa Cruz Genomics Institute led by Benedict Paten, professor of biomolecular engineering and a member of the Zoonomia Consortium. The new papers include a study that identified thousands of elements in the human genome that are highly conserved across species, and another showing how this information could make it easier to find genetic changes that increase disease risk.
“When we do genome sequencing of humans, it can be difficult to tell which genetic variants are significant,” explained Paten, who is a coauthor of both papers. “If it’s at a highly conserved site in the genome, that’s a good sign the variant may have functional effects and increase the risk of disease.”
The Balto paper also used this approach to characterize genetic variation in Balto compared to modern dogs. Balto and populations of working sled dogs had lower burdens of rare, potentially damaging variation than breed dogs, indicating they represent genetically healthier populations. The researchers also identified protein-altering, evolutionarily constrained variants in Balto in genes related to tissue development, which may represent beneficial adaptations.
“Balto had variants in genes related to things like weight, coordination, joint formation, and skin thickness, which you would expect for a dog bred to run in that environment,” Moon said.
Raised in the kennel of breeder Leonhard Seppala, Balto belonged to a population of small, fast sled dogs imported from Siberia that became known as Siberian huskies. The modern Siberian husky breed, however, is quite different from Balto and from modern sled dogs. In addition to Siberian huskies and Alaskan sled dogs, other living dog lineages that share common ancestry with Balto include Greenland sled dogs, Vietnamese village dogs, and Tibetan mastiffs.
“It’s really interesting to see the evolution of dogs like Balto, even in just the past 100 years,” Moon said. “Balto’s population was different from modern Siberian huskies, which have since been bred for a physical standard, but also from modern working Alaskan sled dogs.”
One interesting trait identified in Balto’s genome is a better ability to digest starch compared to wolves and Greenland sled dogs (an isolated population), but not as good as modern dogs, which easily digest starchy foods.
Researchers were also able to use Balto’s genome to reconstruct his physical appearance, including his stature and coat color, in more detail than even historic photos could reveal.
“This project gives everyone an idea of what’s starting to be possible as more high-quality genomes become available to compare,” Moon said. “It’s an exciting moment because these are things we haven’t done before. I feel like an explorer, and once again Balto is leading the way.”
Another paper from Shapiro’s lab and the Zoonomia Consortium, in collaboration with the San Diego Zoo Wildlife Alliance and other institutions, used genomics to predict which mammal species are more likely to face extinction. Genomic analysis can reveal evidence of inbreeding and other indicators of a population on the brink. Although ecological data provide the best predictors of extinction risk, genomics can help identify species that need more attention.
“In conservation, there are more species that need attention than we have time or resources to study, and it turns out that just having one good DNA sample can be enough for us to say either they’re probably alright, or now we need to focus on this species,” Shapiro said.
In addition to Moon and Shapiro, the coauthors of the Balto study include Heather Huson and Krishnamoorthy Srikanth at Cornell University; Kathleen Morrill, Xue Li, and Elinor Karlsson at UMass Chan Medical School; Ming-Shan Wang at UC Santa Cruz; Kerstin Lindblad-Toh at the Broad Institute of Harvard and MIT; Gavin Svenson at the Cleveland Museum of Natural History; and the Zoonomia Consortium. This work was supported by the National Institutes of Health and the Siberian Husky Club of America.